 EN
EN  PT-BR
PT-BR FR
FR ES
ES DE
DE
According to the European Union or Food and Drug administration ( United States) before putting your medical device on the market, you must:
- Prepare a medical device technical file, aka technical documentation or Device Master record, to show proof that the medical device complies with the general health and safety requirements
- Ensure that the technical documentation is made available to the market surveillance authorities as soon as the medical device is placed on the market.
- Keep the technical documentation for 10 years from the date the medical device is released on the market (except specified otherwise).
In this context, a medical device is any instrument, appliance, software, implant, reagent, or material intended to be used alone or in combination for medical reasons.
In this article, you’ll learn (step by step) how to write technical documentation for medical devices.
Things to note: All technical data can be compiled in any of the official languages of the European Union, but English is preferable. The documentation should be submitted in electronic or hard copy or a combination of both. The preferred document format is PDF files with bookmarks to easily locate specific content.
Now, let’s get right into it.
What is Medical Device Documentation, and who is it for?
A medical device technical file, aka technical documentation or Device Master record, is a collection of documents that shows proof that a medical device complies with the GSPR or Medical Device Reporting (MDR) regulation (21 CFR Part 803) (General Health and Safety Requirements), such as the EU MDR Annex I (European Union Medical Device Regulation).
Technical files are essential for all classes of medical files – Class Is, Im, IIa, IIb, III – except for Class I devices, which aren’t sterile and don’t have a measuring function. The files must contain relevant medical device information, including its design, manufacture, and operation. This is needed to justify and support an EU declaration of conformity (See Annex IV of the MDR 2017/745). Technical file documentation is a requirement to have a CE marking affixed on the medical device.
Good to know: CE marking shows that the manufacturer has assessed a device and is declared to meet the safety, health, and environmental protection requirements.
Annex II and III of the MDR 2017/745 go in-depth into what every technical documentation must contain and what it mustn’t, we’d touch on that as we go deeper in the article.
- Annex II – Technical Documentation
- Annex III – Technical Documentation on Post-market Surveillance
- Annex IV – EU declaration of conformity
Importance of Medical Device Documentation
Let’s start with the most important reason. Medical device technical documentation is important because it keeps the end users/patients safe.
1. Clear documentation saves lives and prevents injuries
A medical device is so delicate and important because lives depend on it. All it takes is one error, one misdiagnosis, and someone could lose their life. So it’s pertinent that manufacturers document their devices meticulously to show and prove to the required bodies that their medical device meets all the safety requirements (Annex II and III) before it goes to the market.
2. Communication across manufacturers, healthcare providers, and patients becomes efficient
Technical files serve as a communication tool between manufacturers, healthcare providers, and patients, ensuring enough information to operate the device correctly.
For instance, many individuals own a sphygmomanometer to check blood pressure. The manufacturers of these devices must attach clear instructions on how to use the device so there are no inaccurate measurements.
According to a study data from a randomized crossover trial, “Using the wrong size arm cuff when performing a standard BP measurement can lead to strikingly inaccurate measurements that may lead to a hypertension misdiagnosis.”
All these can be avoided through technical documentation because the manufacturer would show the best ways to use the device, and how to know the accurate size arm cuff to use to get the best result.
3. Improves device usability and accessibility
Just as we said above, technical documentation makes it easier for healthcare providers and patients to use and access a medical device. Especially since there is the quality control and PMS requirements, which include troubleshooting guides, risk assessment, mitigation procedures, etc.
4. Effective training and education of healthcare professionals
Technical documentation for medical devices always contains instructions for use, labeling, and user guides, instruction manuals, or troubleshooting guides. Healthcare professionals say a lab technician or a nurse requires pinpoint training in handling equipment and medical devices. An instruction manual helps them train on how to use the device, and a troubleshooting guide provides quick-fix options if they encounter an issue with device operation. Without proper training, it can lead to huge risks as they deal directly with the patients.
Schedule a demo with one of our experts to take a deeper dive into Document360
Book A Demo
Key Elements of Medical Device Documentation
The MDR clearly defines the elements that need to be in a medical device technical documentation in Annex II and Annex III.
Annex II outlines a list of all the information manufacturers must include in their technical documentation to meet the general safety and performance requirements. Some of the Key elements are
- Device description and specification, including accessories and variants.
- Design and manufacturing information
- Label and packaging information
- Instructions for use in the languages accepted in the states where the device will be sold
- Product verification and validation
- General safety and performance information
- Identify all sites, including suppliers and subcontractors, where design and manufacturing activities are performed.
- Pre-clinical and clinical data
Manufacturers must have a proactive and systematic process for collecting information to develop the post-market surveillance plan.
The post-market surveillance plan should address the collection and utilization of available information concerning serious incidents and any undesirable side effects. It should also address information from trend reporting, relevant specialty or technical literature, feedback, and complaints from users, distributors, and importers.
1. Device Description and specification
The medical device description and specification in the technical documentation is an overview of what the device is about. This section should include:
- Brief description of the product. This includes model names, product costs, configurations, composition, functionality, formulation, variants, and description of the device.
- Photographs, drawings, or schematic diagrams clearly indicate the device’s key parts/components.
- Product History
- Accessories for the device
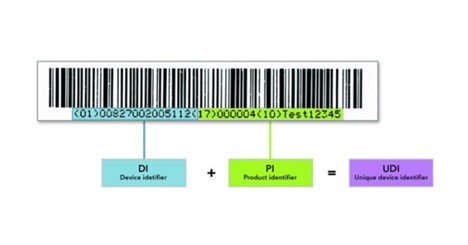
- A basic UDI-DI is assigned to the medical device.
Must Know: A basic UDI-DI (Annex VI Part C) is the primary identifier of a device model. UDI (Unique device identification) is numeric or alphanumeric that identifies the device, lot number, expiration date, and serial number.
UDI-DI is the main key for records in the UDI database and it is used in relevant documentation like the EU declarations of conformity and technical documentation.
2. Instructions for use (IFUs)
Manufacturers need to provide instructions about the intended use of the device, ways to use it, basic principles of operations, intended users, and the indications and contradictions of the product.
Here is high-level information about all the sections required in an IFU:
- Intended medical use: This section should include a piece of brief information to show how your device works. Include the medical condition(s) that your device can diagnose, screen, monitor, prevent, or treat.
- Intended users: This is where you describe your users. Who are they? What level of education? Do they need to have any pre-existing knowledge of the field that your device is used in? Do they need training?
- Describe the ideal patient for your device: Talk about health conditions, age groups, weight range, etc.
- Use environment: This is where you describe the ideal use environment for your device. Does it require users to have a smartphone to use? Does the device only run on one device at a time or on multiple devices? What hardware configuration is required to run your device? What software configuration is required?
- Installation: This is where you talk about how to set up and use the device. Share information about handling and storing the device (if necessary). And give detailed instructions on how and when to perform tests on the device.
- General safety and performance: Use this section to show that your device meets the general safety and performance requirements. Talk about the safety risks, clinical implications, and risk mitigation measures.
- Safety and maintenance: Use this section(s) to discuss safety measures, including troubleshooting. Talk about how users can report malfunctions, lost passwords, data loss, or clinical incidents.
Also Read: Guide to Create Standard Work Instructions for Your Team
3. Labeling
So, it’s a “labeling and instruction for use” section. But we’d just focus on labeling since we’ve talked about IFUs.
This section serves as a communication point between the manufacturer and users to ensure there are no errors with the device and that the users have the necessary information to use the device optimally.
The documents in the labeling section include:
- Label
- User training manuals
- Instructions for use (see above section for more)
4. Manufacturing and technical specifications
The manufacturing and technical specifications of the technical file give insights into the device’s manufacturing process and design. This section should include information about:
- The manufacturing process and specifications, adjuvants, product testing, materials used, quality control procedures, manufacturing process flowcharts including in-process controls, and device specifications.
- The sites, including suppliers and contractors, where the manufacturing processes are performed.
This information is important to show that the device is manufactured best, and meets all the safety and quality requirements.
5. General safety and performance
The information in this section provides evidence that the device conforms to the general safety and performance requirements in MDR Annex I. The information needed in this section includes:
- Mechanical safety reports
- Risk management documentation
- Performance testing reports
- Clinical evaluation reports
6. Quality control procedures
This section usually contains information demonstrating that the device complies with regulations such as ISO 13485. Quality control prevents risk, promotes patient safety, and upholds the quality of medical devices.
For quality control, the technical file needs to have the following information:
- Brief description of the product; intended use and indications for use
- Product labeling and IFUs
- Product specifications
- Manufacturing processes include inspection, packaging, handling, storage, and distribution.
- Technical specifications, including specifications for measuring and monitoring and procedures for product installation and troubleshooting (if applicable).
7. General safety measures and risk management
This section is one of the most important parts of the technical documentation. It provides detailed information on spotting, accessing, and mitigating risks associated with the medical device.
The information that should be included in this section are
- Residual risk evaluation report
- Risk management file including risk analysis, risk management plan, and risk evaluation, implementation, and verification of the risk control measures
- Risk management report
- Risk acceptance statement
- Hazard identification and risk assessment reports
8. Post-market surveillance (PMS) information/Safety monitoring information
This PMS section is so important because it focuses on the device’s safety and performance after it has been placed on the market. This section outlines the manufacturer’s plan for maintaining the device’s safety and performance. You need to take this section seriously. You need to show that you’ve done your due diligence and you have foresight.
The information included in this section is as follows:
- Post-market surveillance plan
- Data analysis and evaluation plan
- Risk assessment and mitigation procedures
- Outline of how you plan to collect data
- Information regarding communication with authorities
Also Read: Product Requirements Document: Benefits, Tips & Examples
Best Practices for writing instructions for Medical Device documentation
Now let’s move on to the best practices you should follow when writing your medical device technical documentation.
1. Prioritize clarity and understanding
There’s nothing worse than writing technical documentation that no one can read and understand. Ever heard of KISS? It’s a popular programming acronym that stands for Keep It Simple, Stupid.
When it comes to medical device regulations, the bar has been raised with the MDR 2017/745, so the regulatory bodies or auditors are out for business. So make sure that every document in your technical file is clear, concise, and understandable. Pay special attention to:
- The clinical data
- Your device’s intended purpose and indication of use
- Labeling
- Performance testing
- Benefit-risk analysis
- Residual risks
- Post-market surveillance plan
2. Provide visual aids for complex concepts
It’s easier to explain certain concepts with visuals than with words. For instance, in the device description and specification section, instead of just talking about the device, you can add images to show how it looks.
You can also use images to demonstrate the process of using the device so the auditor can understand better.
3. Follow content hierarchy for easy navigation
Make sure that your documentation follows the right structure (as seen in Annex II and II) so it can be easily navigated and scanned by the auditors. Here is the recommended structure of a technical file:
- Device description and specification, including accessories and variants.
- Device description and specification
- Reference to previous and similar generations of the device
- Labeling and instructions for use
- Design and manufacturing information
- General safety and performance requirements
- Benefit-risk analysis and risk management
- Product verification and validation
- Pre-clinical and clinical data
- Technical documentation on post-market surveillance
4. Ensure easy accessibility of the documentation
Most people say you can submit the documentation in hard copy or electronic format; we say, “Go with the latter.” Why? It’s easier to access.
Also, the documentation needs to be reviewed periodically and kept up to date. This is why we advise that you go with an AI-powered documentation tool like Document 360 that allows you to easily review the files, make changes, and save.
5. Implement a robust review and approval process
As a manufacturer, there’d be so many other people working with you on the technical files – designers, developers, contractors, etc.
With multiple people involved in the technical documentation process, you need a platform that allows you to collaborate with your partners by allowing them to be able to review your files, comment on them, and approve or disapprove some documents.
6. Ensure adherence to regulations
The medical device industry is highly regulated. That’s why you need to make sure that your technical files adhere to the rules. And keep in mind the more risk a medical device has, the more regulations it must adhere to. So follow the rules to the T to avoid complications.
Also Read: Create Standard Operating Procedures for Clinical Practice
AI-powered documentation tools for interactive and enhanced medical device documentation
The medical device industry is a competitive market that is highly regulated. So you can’t get away with doing anything manually – you need a tool like Document360, that is designed to help manufacturers like you stay compliant with regulations.
Check out our video, AI-Powered Healthcare Knowledge Base:
Why Document360?
With Document360, you can create documents with a clear structure and hierarchy, making it easier for the users to find the information they need without going through any friction points. The AI Concierge, Eddy helps you find the most accurate information faster than ever.
Also, you can easily make changes to your documents. This is very important because your medical device technical documentation needs to be reviewed periodically and kept up-to-date. And the good thing is, you don’t have to do it manually. Once you make a change to any part of your document, it gets reflected everywhere the information is reused. By using Document360, you can make quick changes and stay compliant.
Remember how we said visual aids are better than content for context? With Document360, it’s easier to add images, screenshots, and drawings to your user guides, manuals, and IFUs.
Wrapping Up
So we’ve gotten to the end of this guide. We hope it helps you to kickstart your technical documentation process. It’s not easy to stay compliant and adhere to all the regulations and requirements with all the documentation you have to create. But it can be easier with Document360.
See how Document360 can help you manage your technical documentation more efficiently by booking a demo.

